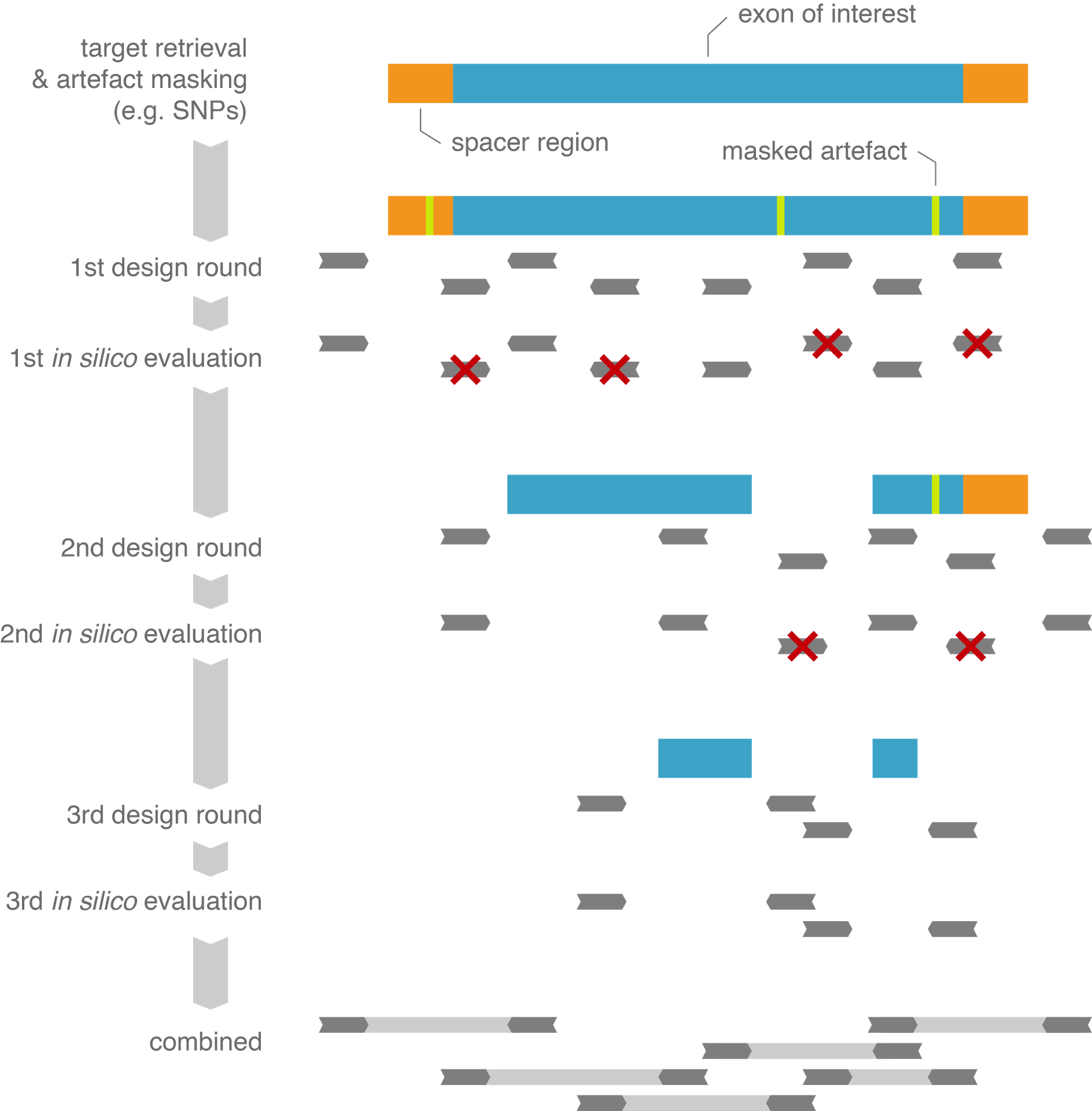
Assay selectivity
In our paper "Single-nucleotide polymorphisms and other mismatches reduce performance of quantitative PCR assays" (Clinical Chemistry, 2013 ), we determined the impact of nucleotide mismatches in primer annealing sites on PCR efficiency.
Various features such as single nucleotide polymorphisms (SNP) and secondary structures in primer annealing sites, are known to have a negative effect on amplification efficiency (see "Evaluation of the impact of single nucleotide polymorphisms and primer mismatches on quantitative PCR" by Boyle et al - 2009 - "Rapid detection of VHL exon deletions using real-time quantitative PCR" by Hoebeeck et al. - 2005 and "Single-nucleotide polymorphisms and other mismatches reduce performance of quantitative PCR assays" by Lefever et al. - 2013 ). Exclusion of these features from primer annealing sites is essential, since these can hamper proper hybridization of a primer to its target sequence. This may result in allelic drop out and reduced amplification.
In addition, assay aspecificity titrates essential components of the PCR mix (e.g. dNTPs) away from the target of interest, resulting in an underrepresentation of the specific amplicon in the end product and potentially leading to non-equimolar end concentrations across PCR reactions requiring normalization prior to library preparation and sequencing.
Our state-of-the-art primer design pipeline takes into account all this and tries to generate assays having as little of these features present in their primer annealing site. Over 94% of our primers are free of SNPs, while for the remaining assays (5.99%), the number of SNPs is limited to one in over 92% of the assays (Figure 1). In these latter cases, the SNPs are located outside the last 5 nucleotides (3’ end) in 85% of the assays to further minimize the effect on amplification efficiency.
As an integral part of our primer design engine, the potential of each candidate assay to generate non-specific (off-target) products, is assessed by aligning the primer sequences to the genome or transcriptome while allowing up to three mismatches per primer annealing site (and taking into account the maximum allowed product length).
By collecting all possible annealing sites, both perfect and imperfect (non-specific) ones and tracking the position of mismatches in them, a score depicting the amplification potential of each site can be calculated.
This strategy enables the selection of the best assays - i.e. guaranteeing no or minimal off-target amplification - and allows our customers to critically appreciate the quality of a ConfirmPCR assay before using it in the lab.
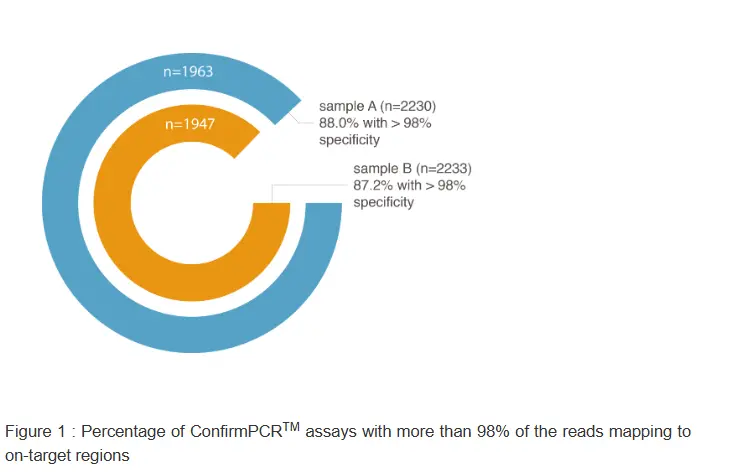
The validity of our in silico specificity score, implemented in our state-of-the-art primer design pipeline, was empirically proven by evaluating almost 2300 assays in two DNA samples (for more details, see the paper in the knowledge center of our website).
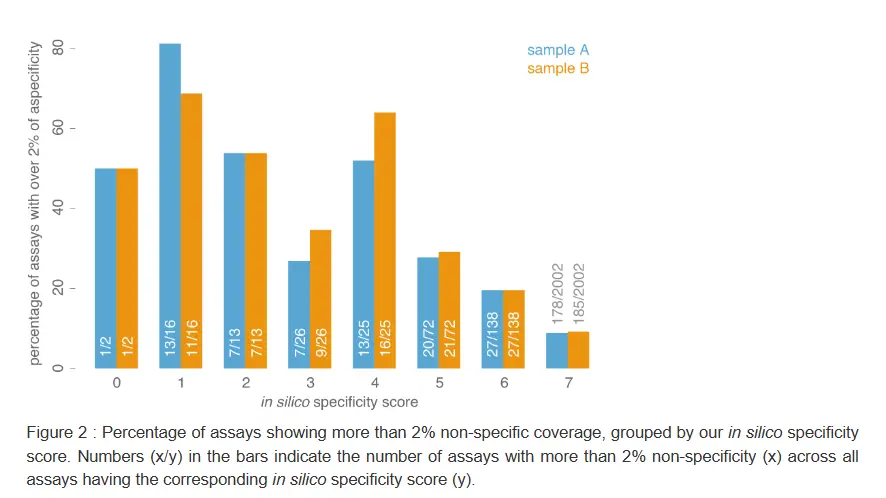
Following standard PCR, the resulting amplicons were sequenced and their on- and off-target coverage determined. The large majority of the assays (88%) demonstrated less than 2% off-target coverage (Figure 1). In addition, a good correlation could be shown between the calculated specificity score (ranging from 1 to 7, low to high degree of specificity) and the percentage of assays having more than 2% aspecific coverage (Figure 2), indicating that assays with higher specificity scores tend to result in less aspecific coverage. Together, these data illustrate once more the quality of our assays and the strength of our in silico specificity assessment workflow and primer design pipeline.